Molecular Modelling
We routinely apply modern computational quantum chemical methods in the course of our work. These are used to investigate reaction mechanisms and origins of various selectivities displayed by processes involving hypervalent iodine.
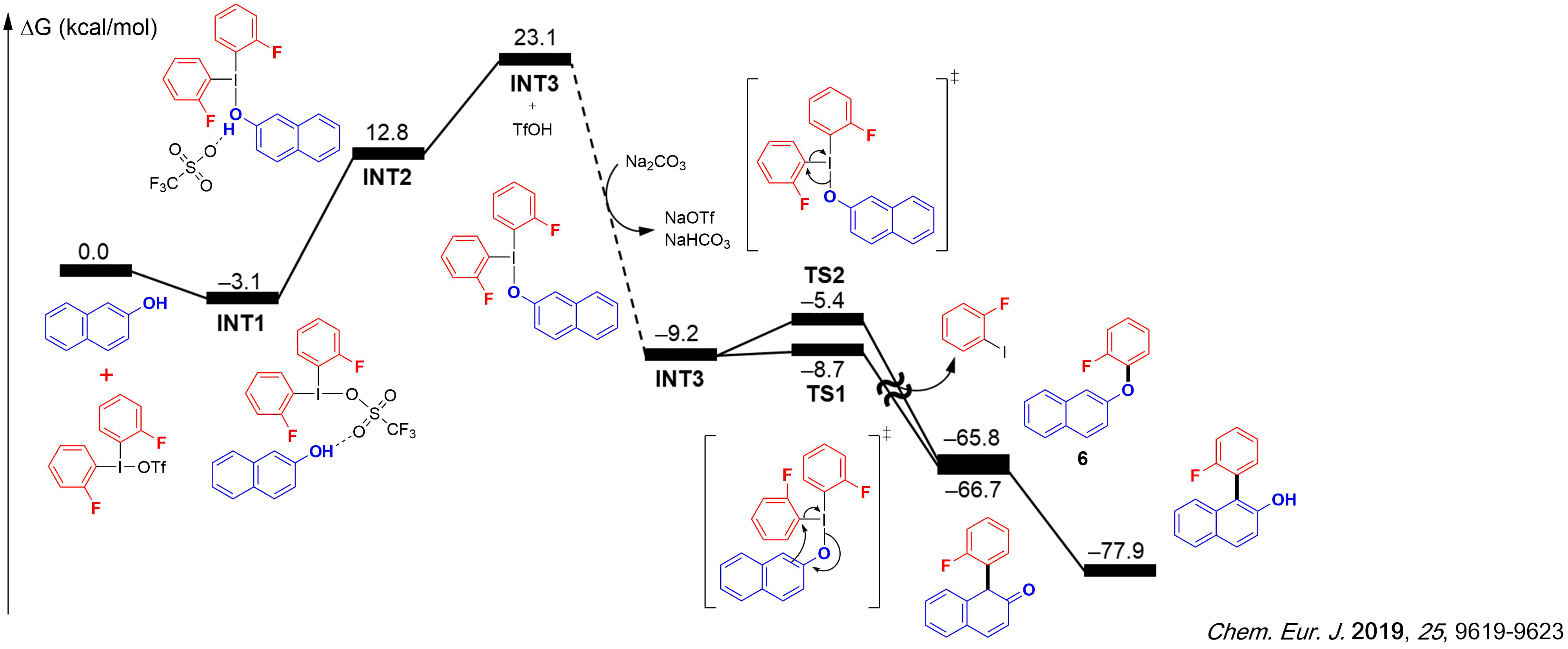
1) We have established computationally that the direct C–H arylation of 2-naphthols with diaryliodonium salts occurs via a dearomatization of the naphthol substrate, followed by a subsequent rearomatization by tautomerization. The reaction follows an inner sphere mechanism, with the incorporation of the naphthol as a ligand for iodine. The selectivity of the process originates from a lower barrier for the C–C bond formation, compared to that for the competing C–O bond formation.
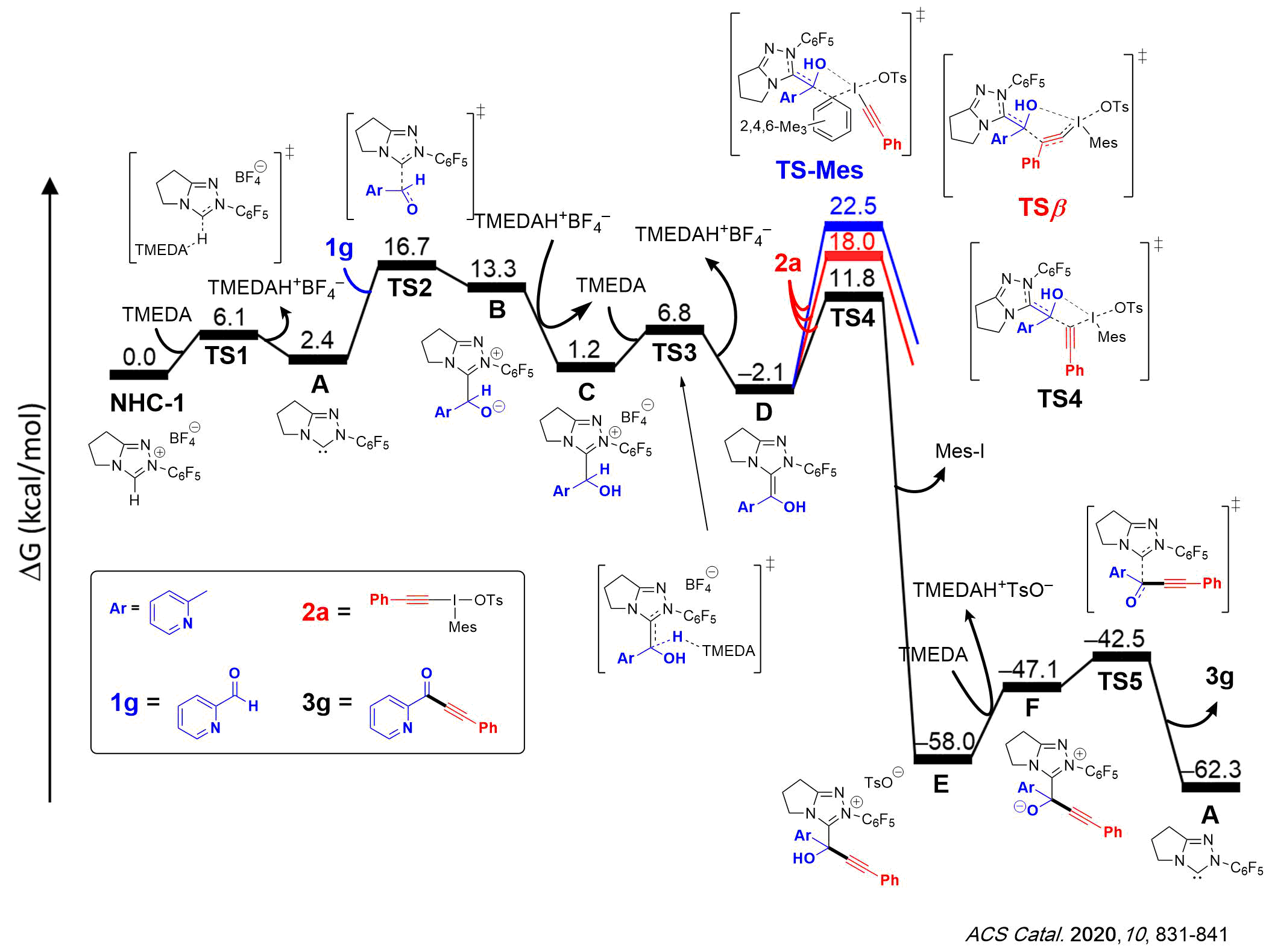
 2) Using computations, we have characterized the mechanism of NHC-catalyzed alkynylation of aldehydes with alkynyliodonium salts. The computations corroborated kinetic measurements, showing that the largest energy span is located between the protonated form of NHC catalyst and the transition state for the carbene attack on the aldehyde substrate. Also, the alkynyl group transfer via a direct substitution of iodine by the Breslow intermediate occurring at α-acetylenic carbon was identified as the preferred pathway, clearly outcompeting the prevalent initial attack of nucleophile at the β-position.
2) Using computations, we have characterized the mechanism of NHC-catalyzed alkynylation of aldehydes with alkynyliodonium salts. The computations corroborated kinetic measurements, showing that the largest energy span is located between the protonated form of NHC catalyst and the transition state for the carbene attack on the aldehyde substrate. Also, the alkynyl group transfer via a direct substitution of iodine by the Breslow intermediate occurring at α-acetylenic carbon was identified as the preferred pathway, clearly outcompeting the prevalent initial attack of nucleophile at the β-position.